New BioRχiv pre-print
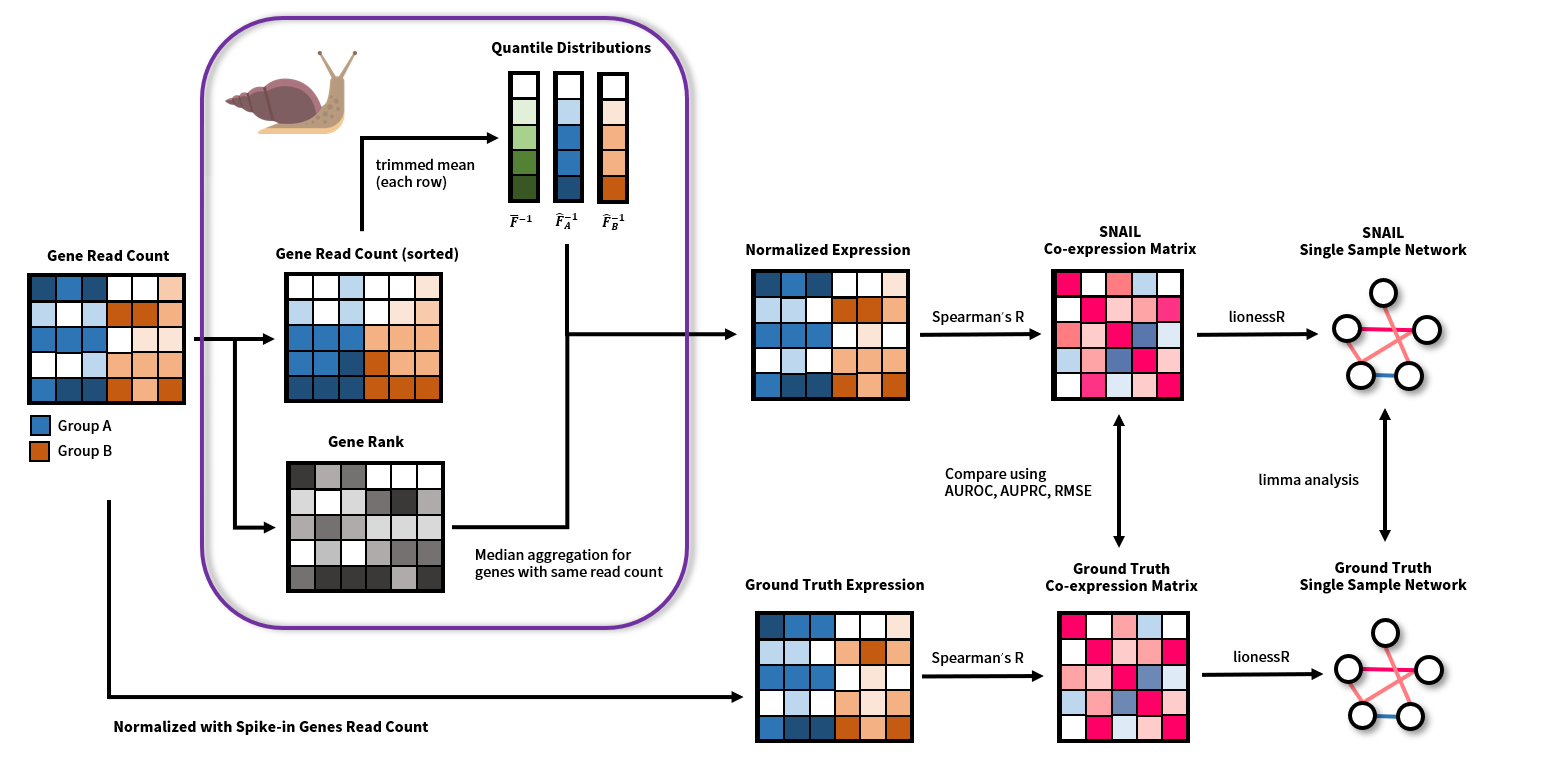
We just posted a new version of our previous BioRχiv pre-print on normalization for network modeling by Ping-Han. Ping-Han found that certain normalization methods, in specific quantile-based methods, can introduce false positive associations in co-expression measurements. However, there methods can be very powerful. Smooth quantile normalization, for example, allows one to normalize heterogeneous data, while keeping global expression differences between different subgroups of samples. Ping-Han therefore developed a new algorithm, called SNAIL, to correct for these false-positive associations, which allows for more precise comparative network analysis in large-scale heterogeneous data. More information on the pre-print can be found here.