New BioRχiv pre-print
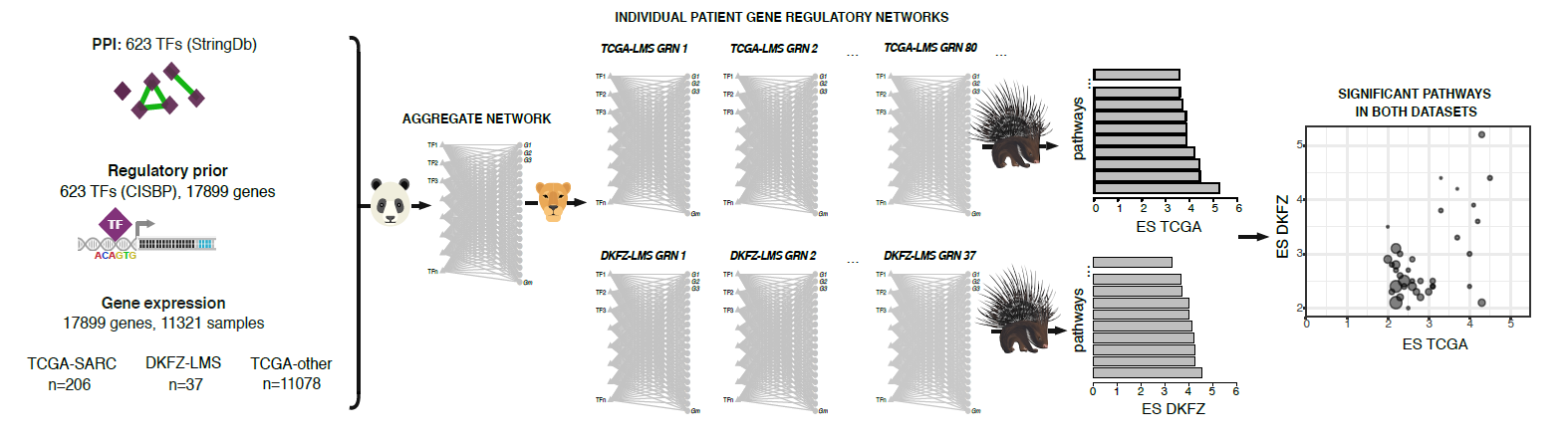
We have posted a new version of our previous BioRχiv pre-print on large-scale network analysis by Tatiana. We modeled networks for individual soft-tissue sarcoma patients and identified large regulatory heterogeneity in leiomyosarcomas. To characterize this regulatory heterogeneity, Tatiana developed a new algorithm, called PORCUPINE. PORCUPINE is a permutation-based approach that uses Principal Components Analysis to identifies pathways that significantly contribute to regulatory heterogeneity in a patient population. We applied the method to an independent leiomyosarcoma dataset and used leiomyosarcoma cell lines to validate identified pathways and genes that drive patient heterogeneity. More information on the pre-print can be found here.