New publication
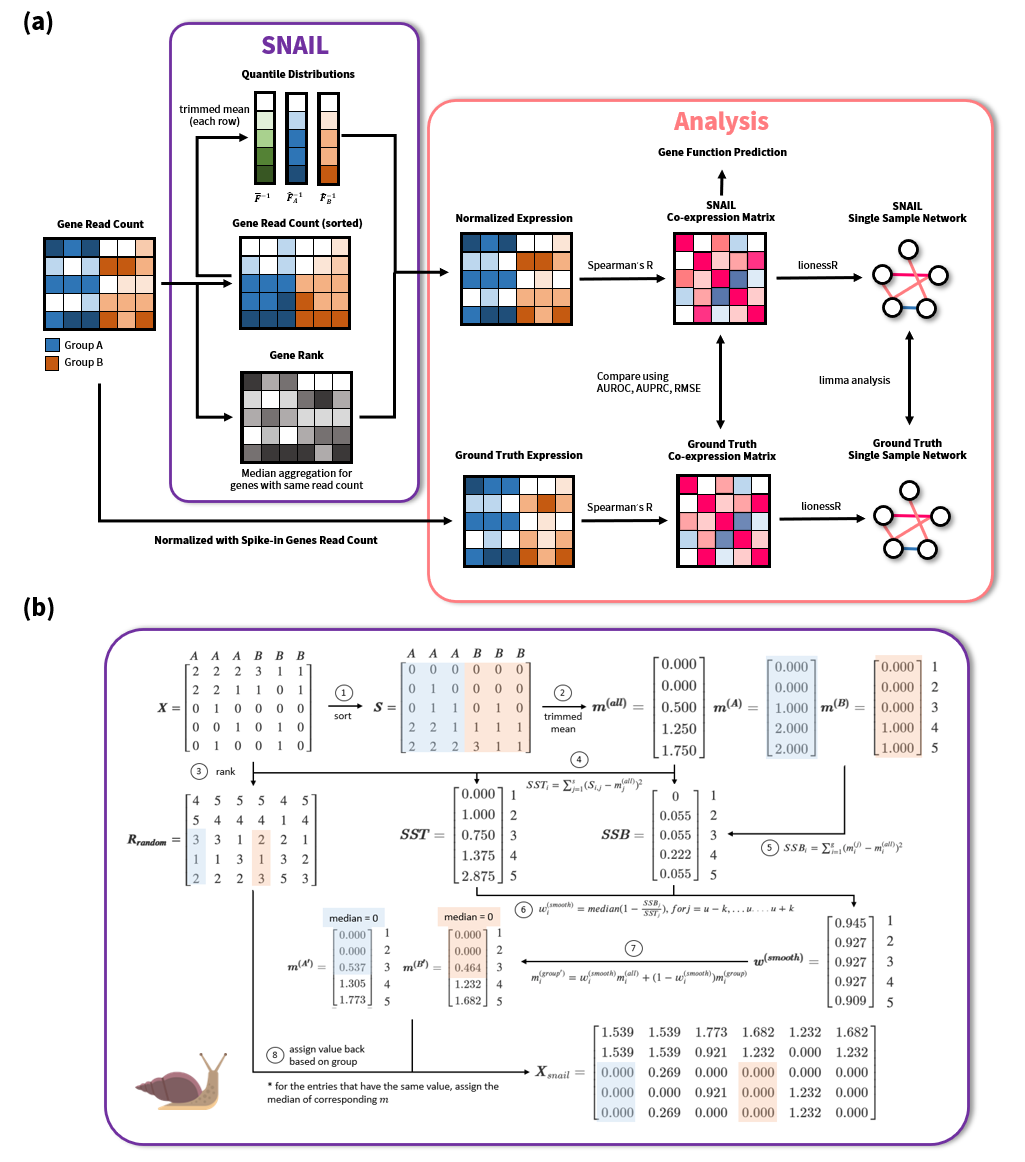
We recently published new work, led by Ping-Han, in Bioinformatics. Ping-Han developed a new normalization method, called SNAIL, which he specifically designed for the pre-processing of large-scale heterogeneous RNA-Seq data prior to network modeling.
- Most normalization methods are designed for downstream analysis of expression, but not co-expression data. We found that normalization can introduce false-positive associations between genes. While this issue can be mitigated by filtering, this leads to information loss, which becomes problematic for heterogeneous datasets. This led us to develop SNAIL.
- SNAIL is specifically designed to normalize RNA-Seq data prior to estimating associations or network inference. The method removes technical variability, while maintaining global differences in expression for samples with different biological attributes. It effectively removes false-positive associations, without the need for filtering. Analyzing data from GTEx and ENCODE, we demonstrate that using SNAIL aids in the detection of, for example, sample-specific network edges, hub genes, and gene function prediction.
- Heterogeneous datasets with increasing numbers of samples and conditions are continuously being generated. We hope that SNAIL will contribute to more precise analyses of such data with association or network-based approaches.
- For more information, please see the publications section.